當將PDB文件上傳到服務器後會給出3個輸出文件:
file.itp為可單獨使用的拓撲文件, file.rtp為可用於b2gmx的拓撲文件, lack.itp文件中定義了缺失的力場參數. 此外, 還會給出所有的輸出文件, 日誌文件和控制台輸出. 讓我們以上面的分子為例來說明生成拓撲的細節.處理步驟
第一步: 程序輸出內部統計的結果
Input file format: Protein Data Bank.
Forcefield OPLS-AA was found in database.
Description: Optimized Potential for Liquid Simulation. All-atomic variant..
Total statistics:
865 atoms, 314 bonds, 988 angles,
1269 dihedrals, 0 nonbonded parameters.
在控制台輸出中, TPPMKTOP會打印出服務器數據庫的統計訊息, 這些值對應於所選力場在數據庫中的總的原子類型數, 鍵結相互作用參數的數目.
第二步: 讀入並處理輸入的化學結構
控制台輸出
Atoms: [.......................................]
日誌文件中會輸出下列信息
Trying to read structure from 'file.pdb'.
LIG - C: 1(1) [ -1.409, 2.132, -0.108] QMname: C
LIG - O: 2(2) [ -2.614, 2.502, -0.908] QMname: O
LIG - H: 3(3) [ -0.928, 3.050, 0.331] QMname: H
LIG - C: 4(4) [ -2.042, 1.200, 1.158] QMname: C
...
從這些輸出你可以檢查原子名稱, 坐標, 元素名稱(非常重要!), 檢查TPPMKTOP是否處理正確.
第三步: 構建原子間的共價連接矩陣
原子間的共價連接矩陣表示哪些原子之間有共價鍵相連, 但這不會在網站中輸出. 這一步是通過調用
openbabel外部過程完成的. 你可以通過將PDB文件轉換為SMILES格式來檢查這一步是否正確完成了.第四步: SMARTS匹配
構建完共價連接矩陣後, 會啟動SMARTS匹配過程, 會在輸出中顯示
3 queries proceeded on database
Calculating scores for every atom.. finished!
Patterns are loading. Please wait.. finished.
Starting SMART-fit.
Patterns checked: 296.
同時在網站中會列出所有匹配的SMARTS模式
Starting curious SMART-fitting procedure.
Loading patterns from database...OK!
[OB] Process PAT: [H,C]C(=O)OC having 5 atoms.
[OB] Process PAT: ClC(Cl)(Cl)Cl having 5 atoms.
[OB] Process PAT: Cl[CX4;$(C(Cl)(Cl)(Cl)Cl)] having 2 atoms.
[OB] Process PAT: [+NH4] having 1 atoms.
...
TPPMKTOP會試著將數據庫中的每一個模式與分子的化學結構進行匹配, 構建出與每個原子匹配的原子類型, 並進行排序.
第五步: 選擇每個原子的原子類型
根據條件得分優先級, SMARTS匹配過程為每個原子選擇一個原子類型
Starting atom_alig..
Filling map..
Applying scores...
為理解這一過程的細節, 讓我們來看下核心數據庫. TPPMKTOP處理完我們的分子後, 在最終的itp文件中, 1號碳原子指認的原子類型為
acetal opls_193(縮醛). 為什麼? 在SMARTS數據庫中, 有兩個記錄與原子1的化學環境匹配, 第一個為(綠色)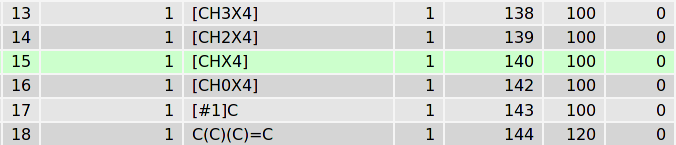
[CHX4]對應於脂肪碳原子, 只含一個氫, 且為4價. 第二個匹配模式(綠色)為
這一記錄意味著
C-O-[CH](C)-O模式的第三個原子應定義為196號原子類型. 這一SMARTS模式對應於下面這種片段: 一個脂肪碳原子一端與氧相連, 另一端依次連著只含一個氫的脂肪碳原子, 連接了兩個碳的氧原子, 一個脂肪碳原子. 選擇是基於兩個匹配模式的條件得分優先級(越大越好): 196號原子類型得分150, 140號得分只有100. 因此, 程序會選擇196號原子類型. 在數據庫內部的記錄中, 196號原子類型對應於opls_193.第六步: 劃分電荷組
在這一步中, 會根據數據庫以及不當二面角(保持平面性或手性)將原子劃分為電荷組, 其算法類似於上面的SMARTS匹配.
CHARGEGROUP patterns are loading. Please wait.. finished.
Starting SMART-fit.
Patterns checked: 8..........
Renumbering CGNR according to human-readable style..finished.
IMPROPER patterns are loading. Please wait.. finished.
Starting SMART-fit.
Patterns checked: 2.
第七步: 生成1-4相互作用
如果所選力場需要明顯地給出1-4相互作用, 則會將它們自動添加到
[ pairs ]部分Generating 1-4 pairs for FF needs..ok.
第八步: 處理缺失力場參數
缺失的力場參數會寫到
lack.itp文件的#define部分, 其係數為零. 通過將參數補充完整, 並將其內容複製到主itp文件的開始部分, 你可以很容易地得到完整的拓撲文件. 對我們的例子而言, 沒有缺失鍵結參數.TPP will write 0 lack parameters to lack.itp.
第九步: 打印電荷
在最後一步, TPPMKTOP會印出體系的總電荷. 如果為系統中的所有原子都正確地指認了原子類型, 那麼這一電荷會等於(或接近)分子的總電荷. 如果仍不相等, 你可以手動修改部分電荷.
Please, correct your charges according to sum: 0.000.
TPPMKTOP finished normally!
你應該檢查一下, 直觀看來, 指認的原子電荷是否與整個化學結構符合. 指認的所有原子類型會在分號後的註釋中列出.
[ atoms ]
1 opls_193 1 LIG C 1 0.300 12.011000 ; C(HCO2): acetal OCHRO
2 opls_186 1 LIG O 2 -0.400 15.999400 ; O: acetal ether
3 opls_194 1 LIG H 1 0.100 1.008000 ; H(CHO2): acetal OCHRO
4 opls_158 1 LIG C 3 0.205 12.011000 ; all-atom C: CH, alcohols
5 opls_169 1 LIG O 4 -0.700 15.999400 ; O: diols
6 opls_170 1 LIG H 5 0.435 1.008000 ; H(O): diols
7 opls_140 1 LIG H 3 0.060 1.008000 ; alkane H.
8 opls_158 1 LIG C 6 0.205 12.011000 ; all-atom C: CH, alcohols
9 opls_169 1 LIG O 7 -0.700 15.999400 ; O: diols
10 opls_170 1 LIG H 8 0.435 1.008000 ; H(O): diols
11 opls_140 1 LIG H 6 0.060 1.008000 ; alkane H.
12 opls_183 1 LIG C 9 0.170 12.011000 ; C(HOR): i-Pr ether, allose
13 opls_185 1 LIG H 9 0.030 1.008000 ; H(COR): alpha H ether
14 opls_183 1 LIG C 10 0.170 12.011000 ; C(HOR): i-Pr ether, allose
15 opls_186 1 LIG O 11 -0.400 15.999400 ; O: acetal ether
16 opls_185 1 LIG H 10 0.030 1.008000 ; H(COR): alpha H ether
17 opls_490 1 LIG C 12 0.190 12.011000 ; C(H2OS) ethyl ester
18 opls_467 1 LIG O 13 -0.330 15.999400 ; AA -OR: ester
19 opls_465 1 LIG C 13 0.510 12.011000 ; AA C: esters - for R on C=O, use #280-#282
20 opls_469 1 LIG H 12 0.030 1.008000 ; methoxy Hs in ester
...
常見問題
引用擴充的力場文件
PPMKTOP擴展了OPLSAA的原子類型, 因此, 有時其給出的拓撲文件中會包含有自定義的原子類型, 如果直接使用的話,
grompp時找不到所需的原子類型, 導致出錯. 解決方法是下載TPPMKTOP擴充的OPLSAA力場. 下載解壓後將其放於類似C:\GMX\GMX5.1.4\share\gromacs\top的目錄下, 並將文件夾更名為oplsaa-erg.ff, 在拓撲文件中#include "oplsaa-erg.ff/forcefield.itp"就可以引用新的力場文件了.異常二面角參數的錯誤
對於OPLSAA力場的
C:\GMX\GMX5.1.4\share\gromacs\top\oplsaa.ff\ffbonded.itp文件, 無論是GROMACS自帶的, 還是TPPMKTOP擴充後的, 都存在異常二面角參數對應的[ dihedraltypes ]段. 原始內容如下[ dihedraltypes ]
; Improper OPLS dihedrals to keep groups planar.
; (OPLS doesnt use impropers for chiral atoms).
; Since these functions are periodic of the form 1-cos(2*x), they are actually
; implemented as proper dihedrals [1+cos(2*x+180)] for the moment,
; to keep things compatible.
; The defines are used in ffoplsaa.rtp or directly in your .top file.
; O?-C -X -Y improper torsion. C can be C_2 or C_3 too.
#define improper_O_C_X_Y 180.0 43.93200 2
; X-NO-ON-NO improper torsion.
#define improper_X_NO_ON_NO 180.0 43.93200 2
; N2-X-N2-N2 improper torsion.
#define improper_N2_X_N2_N2 180.0 43.93200 2
; Z -N?-X -Y improper torsion
#define improper_Z_N_X_Y 180.0 4.18400 2
; Z -CM-X -Y improper torsion. CM can be C= too.
#define improper_Z_CM_X_Y 180.0 62.76000 2
; Z -CA-X -Y improper torsion. CA is any ring carbon (CA,CB,CN,CV,CW,CR,CK,CQ,CS,C*)
#define improper_Z_CA_X_Y 180.0 4.60240 2
這裡定義了一些異常二面角類型的勢能參數, 但沒有給出函數類型參數, 因此如果在拓撲文件中直接使用
1 2 3 4 improper_O_C_X_Y這樣的形式來定義異常二面角, grompp時就會出錯, 給出invalid dihedral type 180的錯誤. 根據GROMACS手冊異常二面角的說明, 這些異常二面角的函數類型應該為4(也可以使用1, 二者沒有區別), 所有隻要在拓撲文件中所有類似i j k l improper_O_C_X_Y的地方增加函數類型, 改為i j k l 4 improper_O_C_X_Y或i j k l 1 improper_O_C_X_Y即可. 不建議直接修改原始的力場文件, 因為這些參數定義會用於氨基酸的二面角, 修改原始力場文件後會導致pdb2gmx生成的蛋白拓撲文件錯誤.
以前的做法
grompp雖然可以成功, 但生成的拓撲文件是錯誤的.參考
https://jerkwin.github.io/2015/12/13/TPPMKTOP-OPLS-AA全原子力场的GROMACS拓扑文件生成器/
http://bbs.keinsci.com/thread-5799-1-1.html
tppmktop單機版編譯
- 編譯需要提前安裝
libmysql++-dev - 需要提前安裝
openbabel,最好安裝在系統默認的文件夾,否則會遭遇pkg-config問題 make的時候報錯,提示和libmysql相關的問題,但是能夠在安裝文件夾的src/programs/路徑發現,tpprenum已經編譯好了,但是tppmktop無法通過編譯。
解決方案: 將下列g++參數中,-lmysqlpp選項挪到最後即可通過編譯。這樣就能得到能夠運行的tpprenum和tppmktop1g++ -std=c++11 -g -O2 -lmysqlpp -o programs/tppmktop programs/tppmktop.o global.o db_scanner.o dbinfo.o smartfit.o db_bonds.o topio.o strutil.o tppnames.o -lopenbabel -lboost_program_options原因:Move the-lflags andmysql_configcommands to the end of the command line. POSIX linkers look at the command line right-to-left: the rightmost object should depend on nothing else, the next to rightmost should depend on either nothing, or only on what is in the rightmost object, etc. The leftmost objects should be those that depend on everything to the right. Since there’s nothing to the right of your program modules, the linker doesn’t see anything it can link AWIWorldServer to. (Except for default libraries like libc, that is, which is always to the right of everything else.)
Your library order is otherwise correct: least generic on the left, most generic on the right.
參考
http://howiedlut.top/tppmktop-compiling/
沒有留言:
張貼留言